








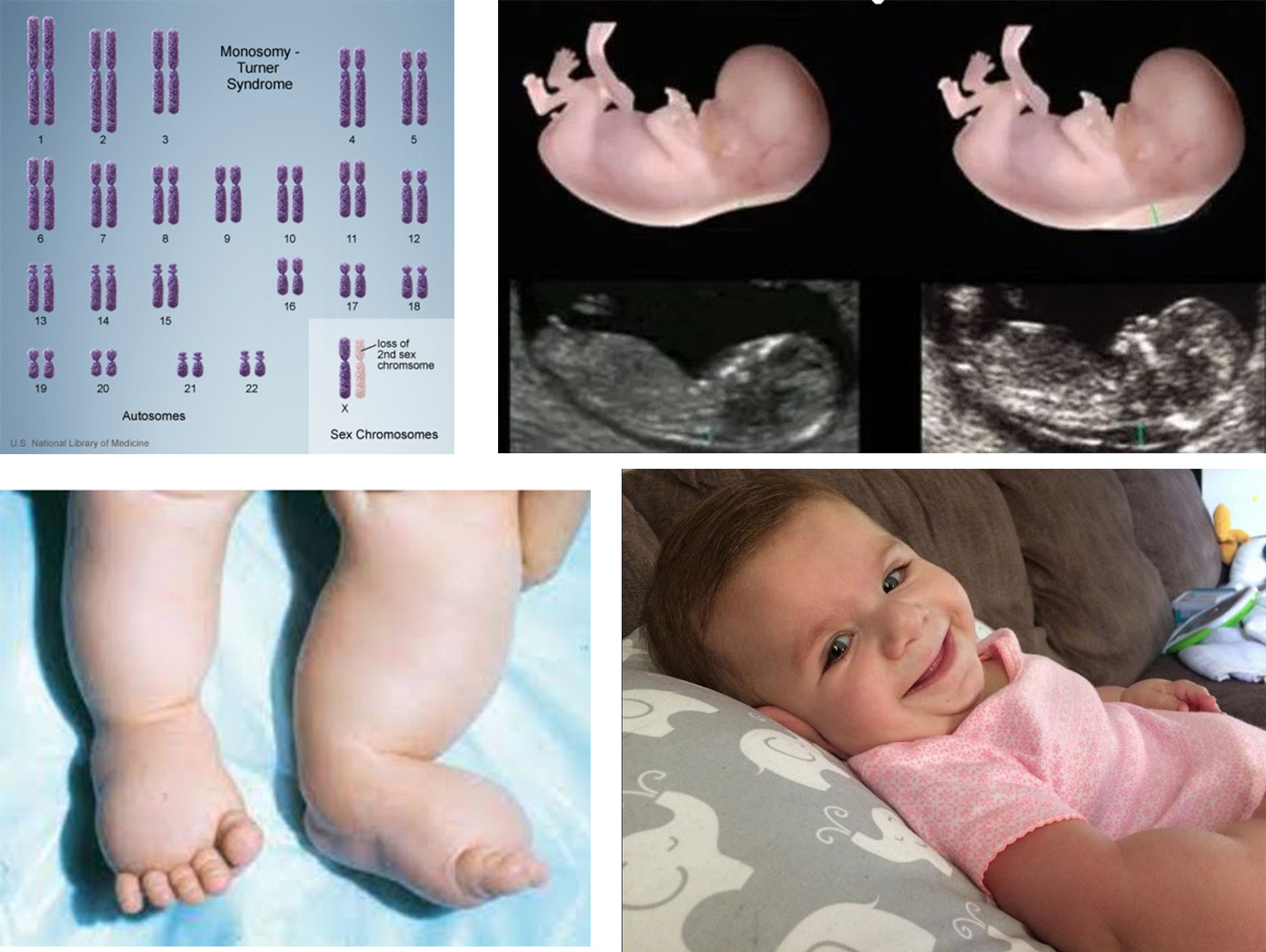
Características del síndrome
Bautizado, como es costumbre, por el apellido de su descubridor (el endocrinólogo estadounidense Henry H. Turner), quien lo describió por primera vez en 1938, también se lo conoce como Síndrome de Bonnevie-Ullrich, Disgenesia Gonadal o Monosomía X. Recién en 1959 el citogenetista británico Dr. Charles Edmund Ford estableció su base genética, cuando descubrió que se trata de una anomalía que solamente afecta a las mujeres y que consiste en la pérdida total o en el reordenamiento estructural de uno de los cromosomas X de los dos que presenta el sexo femenino.
Se trata de una enfermedad rara o poco frecuente porque las estimaciones hablan de que se presenta en 1 de cada 2.500 a 1 en cada 8.000 nacidas vivas, mientras que la Organización Mundial de la Salud indica que una condición entra en tal clasificación cuando su incidencia es menor a 5 de cada 10.000 nacimientos. La diferencia en la consideración podría deberse a que no existen estadísticas oficiales al respecto y a que, como se verá, algunos casos presentan signos tan leves que no se detectan sino incidentalmente. También puede incidir en la diferencia que los casos que se descubren muy tardíamente no suelen entrar en la consideración.
Normalmente, los seres humanos heredamos 23 pares de cromosomas, la mitad de la parte materna y la otra de la paterna. Hay 22 de ellos que se numeran, mientras que el vigésimotercero es el que determina el sexo biológico, XY para varones, XX para mujeres.
Cuando se porta una sola copia en lugar de dos de alguno de ellos se denomina monosomía. En lo que respecta al Síndrome de Turner es la única viable, ya que en los demás casos no se llega al nacimiento, o si este se produce, la sobrevida no pasa de algunas horas o unos pocos días. Ello se debe a que el X restante es capaz de asumir algunas de las funciones del faltante, lo que no ocurre con los otros veintidós. La otra excepción la constituye el llamado Síndrome del Maullido del Gato, pero el mismo no implica que el sujeto carezca de una de las dos copias del cromosoma 5, sino que a una de ellas le falta una parte, es decir que no es sino una monosomía parcial y no total.
En el 50% de los casos la mujer carece de uno de los X (síndrome clásico), mientras que en el resto obedece a reordenamientos estructurales, en los cuales, en lugar de presentar dos brazos largos y dos cortos, en este cromosoma aparecen o unos u otros, perdiéndose la otra parte. Más raramente puede faltar uno de los más extensos o de los más pequeños o también, al romperse, se unen los extremos y se fusionan, conformándose en la forma de un anillo.
Una cuestión a destacar es que, aunque existan algunas evidencias de transmisión materna del síndrome, ello es sumamente raro, ya que la alteración genética se produce de novo en casi todas las pacientes, es decir, sin que existan antecedentes previos, sino que la misma puede realizarse de dos maneras, sea en la disyunción o posteriormente.
La disyunción es el proceso mediante el que los 46 cromosomas paternos y maternos se dividen en dos para que, al juntarse, cada individuo tenga una copia de cada progenitor. Puede suceder que exista una falla por la cual uno de los dos no se separe, mientras que el otro quede huérfano. Si los óvulos o los espermatozoides presentan esta alteración y son los que prosperan, pueden ocurrir dos cosas. Una, que el cigoto, la primera célula de la nueva vida, porte solamente una copia del Y, con lo cual el supuesto varón no es viable, o que aparezca con un único cromosoma sexual X, dando como resultado una niña con Síndrome de Turner. Si ello ocurre, todas las células de esta persona portarán la anomalía.
Ahora, cuando el problema sucede en la mitosis, la continuación de la división celular a partir de la célula original, se produce lo que se denomina mosaicismo, que no es otra cosa sino que la replicación de la mutación se da en todas las que se deriven de la fallada, mientras que el resto no presentará alteración alguna. La cantidad de unas y otras estará dada por el momento en que se produzca la diferencia: cuanto más tarde, menor afectación y viceversa.
Otro tanto ocurre en lo que respecta a los reordenamientos estructurales. En estos casos, lo que sucede es que en la división una de las dos células resultantes se produce un error, según el cual una parte no se separa correctamente, sino que queda adosada a la original, por lo que la otra carecerá de ese segmento o región. Nuevamente, si la que prospera es la versión incompleta de un X, aparece el síndrome, en un 6 al 11% de los casos se verifica la presencia de parte de un Y, viable por ser una alteración parcial.
Los síntomas
Los síntomas son variables en ocurrencia y en intensidad, existiendo una tendencia a que sean más graves en aquellas mujeres con un compromiso celular mayor, aunque, como sucede con todos los síndromes genéticos, ello también depende de los órganos que resulten más afectados.
El signo que resulta más evidente luego del nacimiento es que la talla que presentan es menor en aproximadamente 20 cm de lo esperable al llegar a la edad adulta, aunque ya es visible la diferencia desde la primera infancia, mostrando retrasos en el crecimiento.
Otros indicadores de probabilidad visibles son el cuello palmeado (unión amplia de la piel entre el cuello y los hombros que semeja una aleta), hinchazón en las manos (sobre todo en el dorso) y en los pies producto mayormente de linfedemas (acumulación de líquido linfático), baja colocación de las orejas, paladar alto y estrecho, baja implantación del nacimiento del cabello, mientras que se verifica en un altísimo grado (algunos textos llegan al 97%) una desproporción entre las piernas y el torso, lunares de gran tamaño, tórax ancho y plano con forma de escudo, párpados caídos, resecamiento ocular, mandíbula inferior retraída o pequeña, entre los más frecuentes.
Entre los que no son observables a simple vista se encuentran las cardiopalías congénitas (alrededor del 30 al 45% de las pacientes), usualmente debidas a anomalías estructurales del corazón, sobre todo por estenosis (estrechamiento) de la aorta; la hipertensión arterial, presente en algo así como el 60% de ellas, no debidas a otra causa y con tendencia a aumentar a medida que hace lo propio la edad; las alteraciones nefrourológicas en aproximadamente el 25 al 40% de las individuas, con alteraciones en la estructura de los riñones, malformaciones de las vías urinarias o problemas en los vasos sanguíneos del riñón, las que, aunque raramente producen daños graves a dicho órgano, sí son fuente frecuente de infecciones urinarias recurrentes; los problemas metabólicos, dado que del 15 al 30% muestran hipertiroidismo (algunos estudios elevan los casos de Hashimoto hasta al 34%), además de otras de origen autoinmune, así como una frecuencia mayor de diabetes por resistencia a la insulina, con tendencia a obesidad; los problemas digestivos, que pueden conducir a enfermedades como la de Crohn, cuyas consecuencias, además de molestas, implican complicaciones mortales en los casos de mayor gravedad, o la colitis ulcerosa, con derivaciones de moderadas a graves, que incluyen una tendencia más marcada a desarrollar cáncer de colon en estas últimas; los problemas auditivos, con otitis repetidas a lo largo de la infancia debidas a alteraciones en la estructura del oído interno, y también la hipoacusia neurosensorial, que se produce por una lesión en dicha parte del oído o por daño en el nervio que conecta el órgano con el cerebro; en las niñas mayores suele observarse un desarrollo incompleto o con retrasos importantes de la pubertad, con mamas pequeñas y vello púbico disperso, lo que conduce a que casi todas ellas (96 a 99%) sean infértiles; al tiempo que existe una mayor tendencia en estas mujeres que en el resto de presentar otros problemas tales como intolerancia a los carbohidratos (40%), artritis, cataratas, osteoporosis, ausencia de menstruación, resequedad vaginal, escoliosis y más.
En lo concerniente al desarrollo intelectual, se destaca que las portadoras del síndrome se ubican, en general, dentro del promedio, aunque en algunas de ellas, un sector minoritario, se observan algunos déficits en el aprendizaje, lo que tiende a afectar más las áreas que no implican a las palabras, tales como las matemáticas o las relacionadas con las formas gráficas y los dibujos, mientras que se incrementa el riesgo de que aparezcan trastornos por déficit de atención e hiperactividad, así como es posible que desarrollen dificultades para desempeñarse en situaciones sociales, mostrar cuadros de ansiedad y depresión.
Detección
Como sucede con cualquiera síndrome o alteración de la salud, cuanto antes se detecte, mejor. Lo que ocurre en algunos casos es que, sea porque los síntomas son insignificantes o difusos o porque es posible confundirlos con los de otras condiciones, recién se toma conciencia al llegar a la pubertad. Más frecuentemente ello se produce en la primera infancia o algo más tarde, al verificarse la presencia de signos que llevan a la sospecha. Uno de los indicadores que suelen disparar las consultas o que advierten los pediatras son los retrasos en el crecimiento, los que ya en los primeros meses se manifiestan por un menor desarrollo de la talla, usualmente alrededor del 5% de lo esperable, pero no es el único.
Con el desarrollo de nuevas técnicas de exploración mediante imágenes o los avances en las existentes, la detección del Síndrome de Turner es posible realizarla antes del nacimiento, aunque normalmente en un grado de sospecha. En general, dependiendo de lo marcados que sean los signos que presente la monosomía en el embrión, lo que suele corresponderse con el grado de gravedad de los síntomas, será más fácil o más difícil la presunción.
En algunos casos, las ecografías prenatales muestran algunos rasgos que pueden indicar la presencia del síndrome. Ellos suelen consistir en una traslucencia nucal (engrosamiento del pliegue de la nuca) muy aumentada, característica usual también en otros cuadros, como en el Síndrome de Down; un higroma quístico, tumor que se presenta en la cabeza y en el cuello; además de edemas subcutáneos; derrames en las cavidades fetales serosas, espacios vacíos del cuerpo que están rodeados de una membrana interna lubricada; hidropesía, es decir, la retención de líquidos en diversas partes del cuerpo, que pueden aparecer ya en la primera mitad del período gestacional y que cuando son más patentes indican la probabilidad de una afectación importante de la niña por nacer e incluso lleva a una altísima posibilidad de mortalidad intrauterina, la que se ubica en un rango de entre el 80 y el 90%. Se cree que por ello la incidencia de esta monosomía al nacimiento es mucho menor que la existente en el feto, ya que algunos estudios indican que, en realidad, algo así como el 3% de los embriones femeninos concebidos portaría esta mutación y no llegaría a término.
La sonografía, otra denominación de la ecografía, o la ultrasonografía, una técnica que combina imagen y endoscopía, permiten la detección de esta y otras anomalías en el seno materno durante el embarazo. Un estudio con esta herramienta que abarcó a 22.150 embarazadas y halló que 514 de los embriones portaban alguna anomalía genética, mostró que 69 portaban signos del Síndrome de Turner en el primer trimestre, 18 un higroma (29,8%), 8 hidropsia (11,6%), 9 defectos cardíacos (13%, 44% de los cuales consistieron en estrechamiento de la aorta) y en 9 (13%) se observó una translucencia nucal importante, entre otros.
Estos resultados funcionan como alertas, ya que cuando los signos no son muy evidentes, existe no solamente el riesgo de falsos positivos, sino también de que algunos de ellos se deban a otros problemas.
Es por ello que la detección certera implica un segundo paso, que es la corroboración genética o no de la existencia de esta monosomía. Para ello se recurre a técnicas invasivas o a otras no invasivas.
Las invasivas son básicamente dos, la amniocentesis y la biopsia corial. La primera consiste en una punción mediante la cual se extrae líquido amniótico, el fluído que rodea al feto en el vientre materno, para realizar los análisis correspondientes que indiquen la existencia de este síndrome o de cualquier otra alteración genética que se sospeche o la descarte.
La segunda consiste en la extracción de las vellosidades coriales de la placenta, pequeñas salientes de la membrana fetal con forma de dedos, para luego someter las muestras a los análisis correspondientes.
En la amniocentesis existen riesgos de rotura de la bolsa y una probabilidad de aborto cercana al 1% de los casos. Otro tanto ocurre con la biopsia corial, sumado a la posibilidad de sangrado vaginal.
Entre las semanas 10 y 11 comienza a ser detectable este síndrome mediante técnicas no invasivas, las que tienden a tener una eficacia menor que las otras, aunque sin consecuencias ni para la madre ni para la bebé. Ellas consisten en tomar muestras de la sangre materna, la que contiene lo que se denomina ADN fetal libre, es decir que no está contenido en el núcleo de las células maternas, ya que proviene de la placenta, la unión del feto con la circulación sanguínea materna, por lo que se liberan pequeños fragmentos pertenecientes al embrión, los que se separan y se analizan. Sirve para hallar la presencia de una copia extra en algún cromosoma o una faltante en uno de ellos, tal como es el caso del Síndrome de Turner.
Estas técnicas sirven como herramienta de detección, sin que con ellas se complete el diagnóstico, el cual deberá confirmarse o desecharse realizando las pruebas genéticas correspondientes sobre el material genético de la nacida.
Tratamiento
No existe un tratamiento capaz de revertir el problema de base, aunque sí los hay referidos al alivio o la solución de muchos de los problemas asociados, de acuerdo a cómo se presenten en cada una de las pacientes.
Generalmente, los problemas relacionados con la baja estatura se resuelven mediante el suministro de la hormona del crecimiento, la que suele comenzarse alrededor de los 3 años y que ayuda a aumentar la velocidad de crecimiento y a mejorar la talla.
Por su parte, los problemas con los retrasos de la pubertad suelen tratarse con el suministro de estrógenos, procedimiento que se emprende cerca de los 12 años.
Para el resto de las derivaciones deberá recurrirse a distintas especialidades, tales como ginecología, cardiología, urología, otorrinolaringología, gastroenterología, etc., de acuerdo con las características de cada paciente.
La buena noticia es que con la contención familiar y, cuando ello sea necesario, con el apoyo psicológico, más la ayuda médica, estas mujeres pueden desarrollar una vida plena.








{kind=link}